|
| Majzoub Research Group |
Openings
Please contact me if you
are interested in working in the group.
|
|
|
| Popular
Press |
|
|
| Current
Research Projects Past Research Projects Hydrogen Storage: Materials
of current interest for hydrogen storage applications
include complex anionic hydrides such as the class of
materials known as alanates and borohydrides.
Examples include NaAlH4, LiAlH4,
Ca(BH4)2, and LiBH4.
These materials are generally wide gap insulators, and
are very different in their material properties from
interstitial metal hydrides. In a classic
interstitial hydride, the metal alloy composition
remains the same before and after hydrogenation.
For example, the compound LaNi5 reversibly
absorbs hydrogen to become LaNi5H6.
In contrast, the complex hydrides undergo decomposition
reactions. Sodium tetrahydroaluminate reversibly
decomposes and reforms according to the following
reactions:
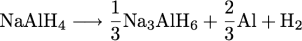 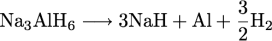 The decomposition reactions yield about 5 wt. % hydrogen, a significant increase over the classic interstitial LaNi5H6, with about 1.4 wt. % H2. Many complex hydrides contain much larger gravimetric hydrogen capacities.  Na and Al are
immiscible, even in the melt. The compound
NaAlH4 exists as an ionic molecular
solid, with AlH4- anions bound
with polar covalent Al-H bonds. Likewise, Na3AlH6,
is composed of AlH63- anions,
charge balanced by three Na+ cations. In-situ single
crystal Raman scattering studies have shown that the
AlH4- anions are stable up to
the melt in NaAlH4 (E.H. Majzoub, V.
Ozolins, K.F. McCarty, Phys. Rev. B, 71, 024118,
2005), limiting the
explanations of enhanced sorption kinetics through
transition-metal "doping" procedures commonly used
for this compound. Our group studies the
structure, lattice dynamics, and thermodynamic
properties of these materials to develop higher
hydrogen capacities and better hydrogen sorption
kinetics.
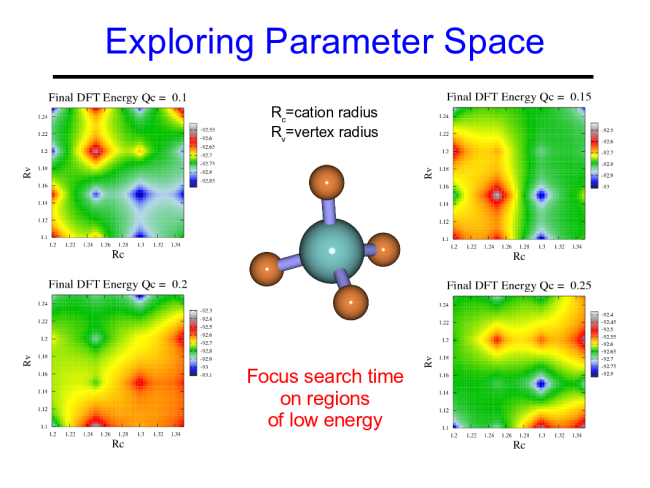
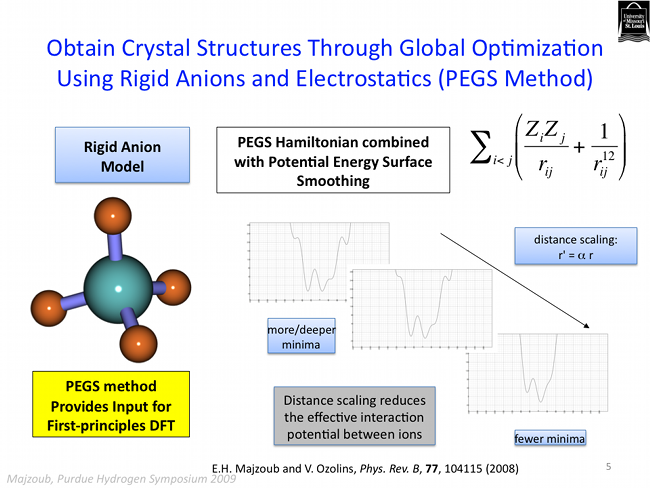
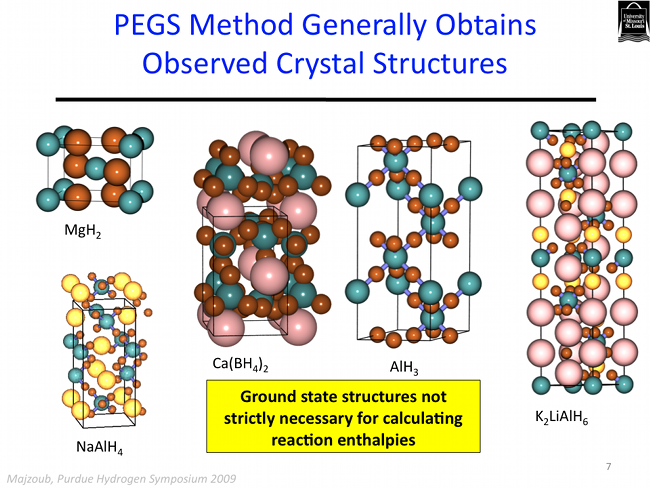
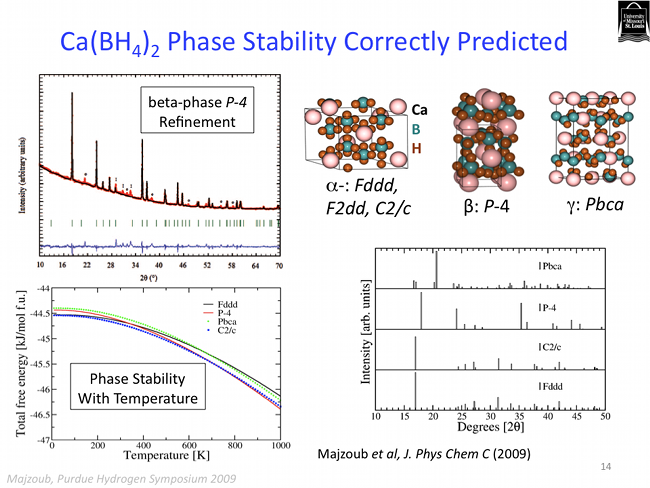
The PEGS method is quite robust. In addition to predicting ground state structues of ionic crystals, it is also able to address the more complicated issue of crystal polymorphs. A polymorph is simply a variation on a crystal structure, and many crystals phase tranform into different structues as a function of temperature, or pressure, for example. Several of the polymorphs of calcium borohydride have been predicted using the PEGS method as shown below.
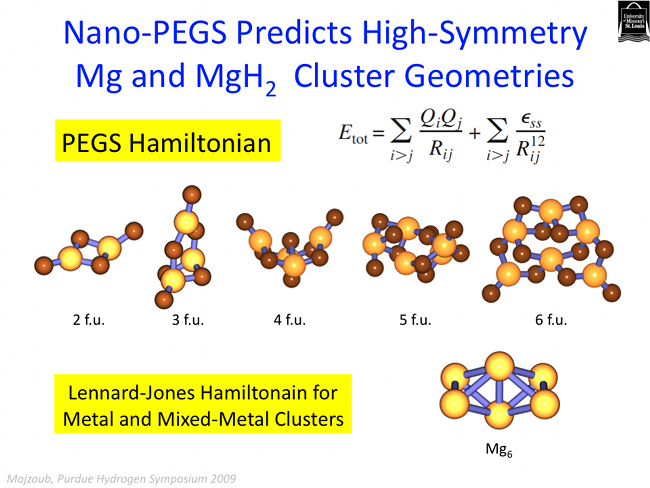
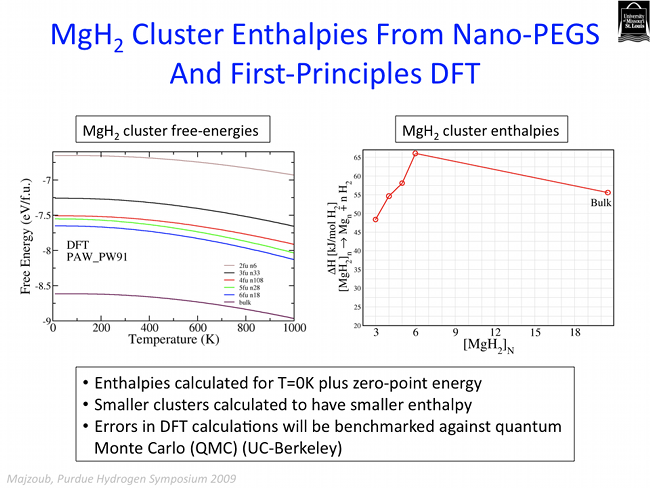
While recent interest has focused on complex metal hydrides such as NaAlH4 and Ca(BH4)2, these compounds are not as easily tunable (as are the interstitial metallic hydrides) through alloying with other metal atoms due to the strongly ionic character of the cohesive energy. However, the complex hydrides are superior on a wt.% hydrogen basis, and are the preferred materials for vehicular transport. In order to address thermodynamic tunability, we investigate these materials at the nano-scale, where the ratio of surface to bulk atoms impacts the energetics. Recent theoretical work by Wagemans et al. [J. Am. Chem. Soc., 127, 16675, 2005] and others indicate that small clusters of MgH2, for example, can significantly lower the desorption enthalpy with respect to bulk. Small metal or hydride clusters may be incorporated into nanoporous frameworks such as block polymer templates, for example, to prevent agglomeration and perhaps even improve tunability through particle/surface interactions.
The figure below shows the total free energy, including entropy, of small clusters of MgH2, calculated using first-principles density functional theory, with structure prototypes generated using the stochastic methods described above. The free energy of small clusters is expectedly larger than that of the bulk and are therefore "destabilized" with respect to the bulk.
Nanocluster hydrides may be housed in a porous framework with controlled size nanopores. We are currently investigating incorporation of small clusters of complex hydrides into these frameworks in collaboration with Sandia National Laboratories.
Surface Enhanced Raman Scattering Substrates: Page under construction 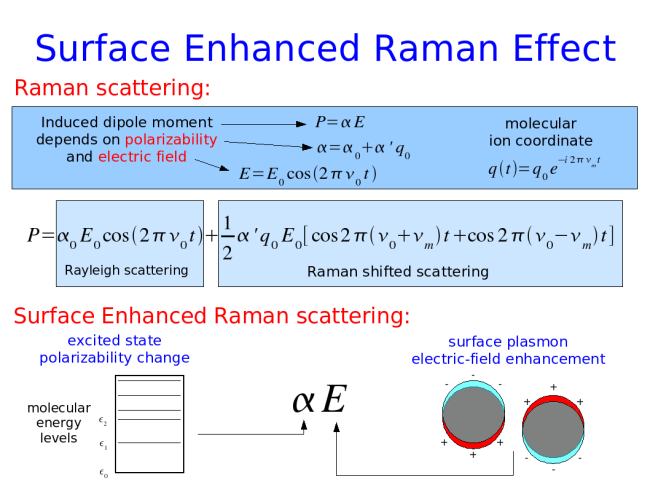 |
|
|
Collaborators
|
| Go back |
| Updated:
07 May 2007 |